Overview
BioTransformer Options
The user can select between 8 different options for metabolism prediction or metabolite identification. The first 5 options allow for a specific coverage of metabolism and are CYP450, EC-Based, Phase II, Gut Microbial, and Environmental Microbial options. Two options allow for a comprehensive coverage of metabolism in the human superorganism and include the allHuman and SuperBio options. The last option is Multi-Step which allows a user to specify the order of individual options.
- CYP450: Select this option for CYP450 metabolism prediction. Cytochromes P450 are a superfamily of enzymes that play a very important role in drug metabolism and usually increases the water solubility of the substrate, converting lipid-soluble compounds to more water-soluble ones, often through oxidation. These enzymes metabolize thousands of endogenous and exogenous compounds. Some metabolize one (or only a few) substrates while others may metabolize multiple substrates. CYP450 metabolism involves most of the reactions in Phase I metabolism, whereby a compound’s reactivity is increased and hydrophilicity is slightly increased.
- EC-Based: EC-Based: Select this option for the prediction of promiscuous metabolism (e.g., glycerolipid metabolism). The Enzyme Commission (EC) assigns numbers to enzymes based on the reactions they catalyze. These enzymes may have multiple substrates, products, and/or reactions, and hence the lack of strict specificity makes them “promiscuous”. This prediction option generates metabolites using reaction rules associated with different enzymes based on the EC numbering scheme.
- Phase II: Select this option for the prediction of major conjugative reactions, including glucuronidation, sulfation, glycine transfer, N-acetyl transfer, and glutathione transfer, amongst others. Phase II reactions strongly increase hydrophilicity to detoxify compounds by enabling renal excretion.
- Gut Microbial: Select this option to predict xenobiotic (i.e., exogenous compounds) metabolism by gut microbial enzymes. The gut microbiota is very important in transforming dietary components, drugs, and industrial chemicals into metabolites, and its chemistry is often distinct from that of the host’s enzymes. Microbiotic enzymatic reactions are typically hydrolytic or reductive in nature, in contrast to host enzymatic reactions being oxidative or conjugative.
- Environmental microbial: Select this option to predict metabolism or degradation of small organic molecules by environmental microbes.
- AllHuman: Select this option to predict small molecule metabolism in the human superorganism. This covers biotransformations occurring both in human tissues as well as the gut microbiota. Each step of the metabolism prediction covers human as well as gut microbial transformations, when applicable.
- SuperBio: SuperBio was originally designed to explore all possible metabolites in the human body for the query compound by running the allHuman module till there are no more novel metabolites generated. However, the SuperBio module currently on our web server runs the allHuman module for only 4 iterations in order to save time and conserve resources so that BioTransformer is available to run queries from more users. To run the original SuperBio module, please download the jar file and run it locally.
- MultiBio: Select this option to create your own biotransformation sequence via any combination of CYP450, EC-Based, Phase II, Gut Microbial, and Environmental Microbial metabolism options. You may choose up to 3 iterations for each option, subject to an overall total of 4.
- AbioticBio: Select this option to make predictions for abiotic transformations including photochemical, chlorination, ozonation reaions, etc.
Web Server Accessibility
The webserver can be freely accessed in two ways: Users can submit queries, and retrieve results both manually and programmatically as described on the next pages.
Fair usage policy
In order to ensure that resources are shared properly, we have limited the number of starting compounds per query to 1. Thus, the tab-separated structural input should have no more than 1 line, and the SD File (SDF) must contain no more than 1 compound. Users who would like to submit more compounds at once or predict more multi-step metabolism are asked to used the command-line executable available here.
Metabolism Prediction
The Input
- The task type (required): The user must select the “Metabolism Prediction” task.
- The BioTransformer option (required): The user can select one of the options depending of the aspect of metabolism that is of interest. The prediction could be specific (CYP450, Phase II, or EC-based), or comprehensive (Gut microbial, Environmental microbial, AllHuman, or Superbio). For a detailed explanation of the different option, please read the Overview page.
-
The structure input (required): The input structures for starting compounds can be submitted in two different ways:
- A tab-separated text: Each line contains either a (preferably isomeric) SMILESstring, or a standard InChI of a starting compound. The structural representation (SMILES or standard InChI) can be preceded by an identifier (e.g. a name, a database ID).
- An SDF file: The SDF can be uploaded, and must contain the exact structure of each starting compound. For each starting compound, the optional identifier should be inserted in the header, just before the corresponding connection table. For more information, please consult the following document.
- The Number of iterations (optional): This determines the maximal number of biotransformation steps that will be predicted.
- The Submission title (optional): This allows the user to specify a title for the query. Please keep in mind that queries are accessible from the web server only through their query ID, as returned by BioTransformer.
The Output
As default, the query output is returned in an HTML format that is displayed on the web interface.

Metabolite Identification
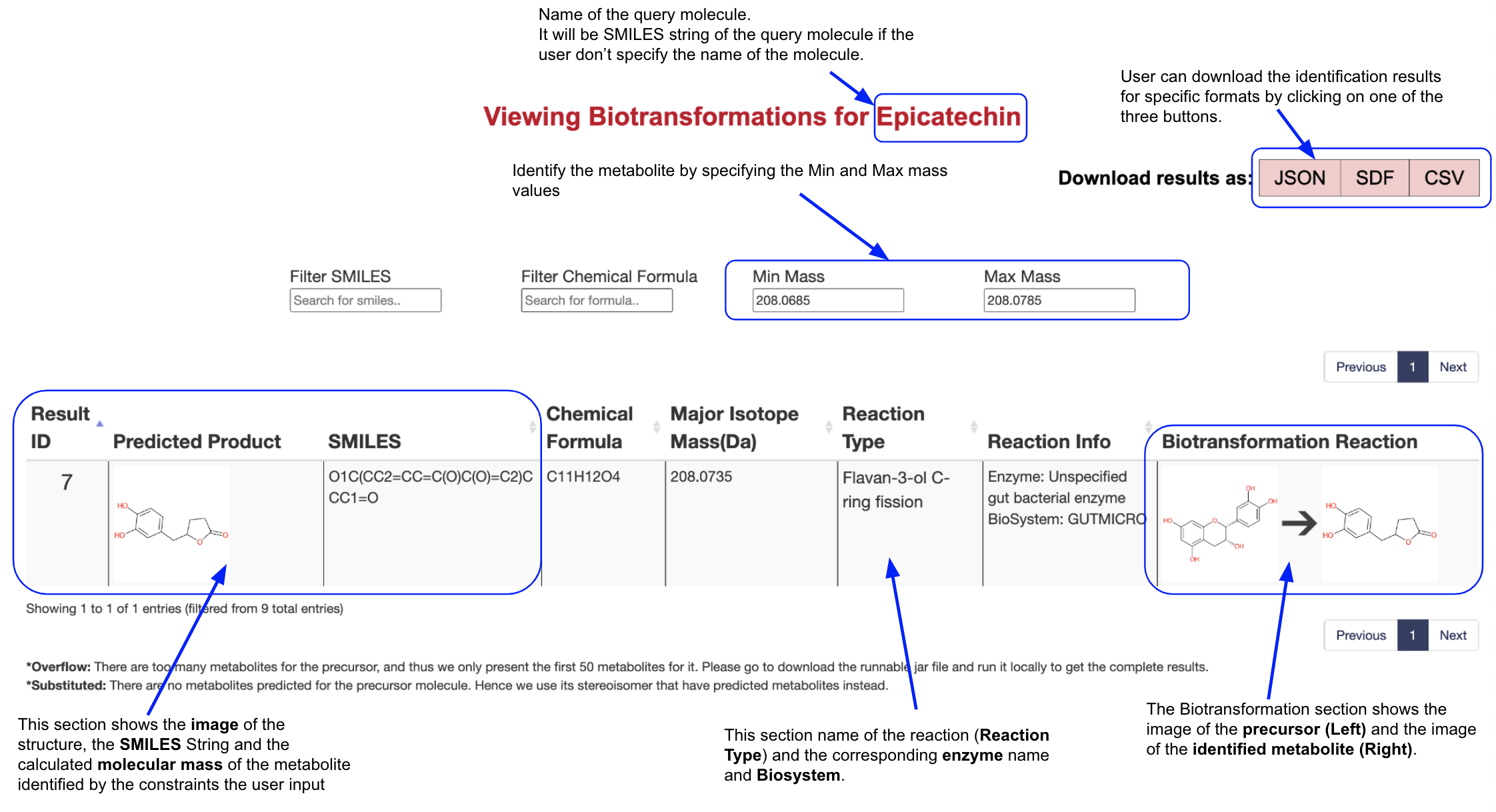
The users now can identify the metabolites of their interest using the Chemical Formula, Min Mass and max Mass filters. For example, if the user wants to identify the metabolites whose mass equals 208.0735 with a tolerance of 0.005 for Epicatechin, they should fill 208.0685 in the Min Mass filter and 208.0785 in the Max Mass filter. The result page will then only present the predicted metabolites that satisfy the conditions.
Programmatic Access
Resources
There is one end-point available to submit queries and retrieve the results from the queries performed:
biotransformer.ca/queries.json
Submit query (POST)
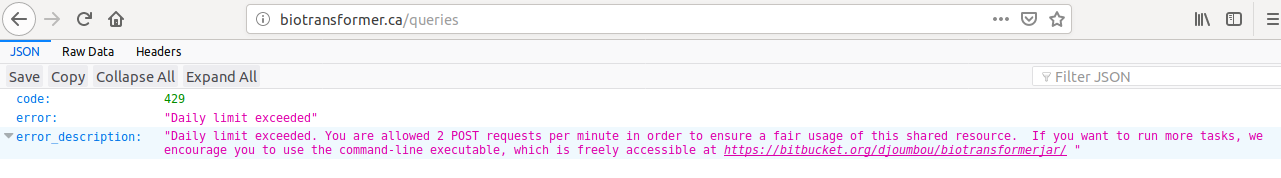
POST: Submit a query to the biotransformer app. In the production server, the number of post requests is limited to 2 per minute. The attributes are listed below. Only one of the parameters ‘query_input’ or ‘fstruc’ must be provided with content.
curl -i -H "Content-Type: application/json" -H "Accept: application/json" http://biotransformer.ca/queries.json
-X POST -d '{"biotransformer_option":{OPTION}, "number_of_steps":1, "query_input":{QUERY_INPUT} ,"task_type":{TASK TYPE}}'
Examples of query submissions are shown below:
curl -i -H "Content-type: application/json" -H "Accept: application/json" http://biotransformer.ca/queries.json
-X POST -d '{"biotransformer_option":"CYP450",
"number_of_steps":1,
"query_input":"acetaminophen\tCC(=O)NC1=CC=C(O)C=C1",
"task_type":"PREDICTION"
}'
Where the json sent is:
If the number of allowed requests is reached in the production server, the user will see the next message:
Retrieve results (GET)
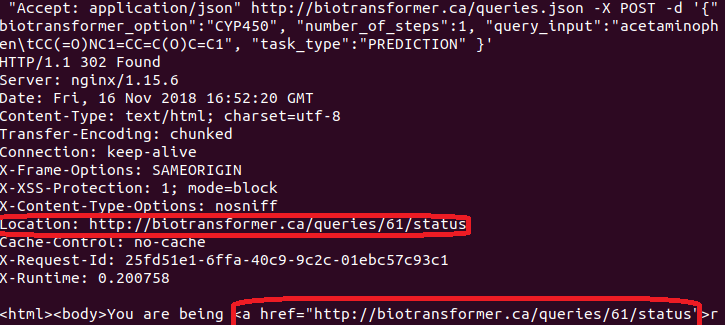
curl -H "Accept: application/json" -X GET http://biotransformer.ca/queries/{QUERY_NUMBER}.json
The query number can be shown in the response from the server when performing a POST request (see image below):
The attributes of the query results for predictions are:
Browser Compliance
| OS | Version | Chrome | Firefox | Microsoft Edge | Safari |
|---|---|---|---|---|---|
| Linux | Ubuntu 18.04.1 lts | 96.0.4464.110-1 | 95.02 | 96.0.1054.62 | N/A |
| MacOS | Monterey 12.0.1 | 96.0.4464.93 | 95.02 | 96.0.1054.62 | 15.1 |
| Windows | 10 | 96.0.4464.110.1 | 95.02 | 96.0.1054.62 | N/A |
Substructure Filter
Example
The user wants to know which metabolites produced by AllHuman metabolism for glucose contains phosphates. He should do following steps:
